

Robust Estimation of Gene Trees
 Gene trees provide the fundamental information in molecular phylogenetics. Originally, a single or a few well curated genes were used for phylogenetic inference. Then, the species tree was assumed to be the same as the estimated gene tree. Today, we often have hundreds or thousands of loci in our datasets. Hence, we can examine comparatively the substitution process for each single locus and ask new research questions, such as: (a) Did this gene evolve under selective pressures? (b) Did this gene evolve under a time-reversible or non-reversible model? (c) Did the substitution process change along the lineages of the tree? (d) Did this gene evolve under a strict molecular clock?
Gene trees provide the fundamental information in molecular phylogenetics. Originally, a single or a few well curated genes were used for phylogenetic inference. Then, the species tree was assumed to be the same as the estimated gene tree. Today, we often have hundreds or thousands of loci in our datasets. Hence, we can examine comparatively the substitution process for each single locus and ask new research questions, such as: (a) Did this gene evolve under selective pressures? (b) Did this gene evolve under a time-reversible or non-reversible model? (c) Did the substitution process change along the lineages of the tree? (d) Did this gene evolve under a strict molecular clock?
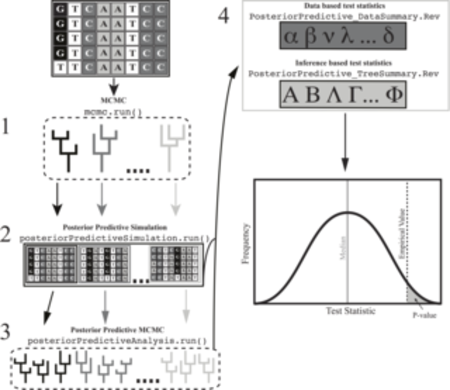
To answer these questions we develop more realistic substitution models in RevBayes. For example, we have designed RevBayes to model heterogeneous substitution process along the branches of a tree which can be used to model variation in GC content among lineages as well as variation in selective pressures along lineages. Additionally, we develop more efficient methods to test for the relative fit a model (Höhna et al. 2017, Bioinformatics) and develop new methods, e.g., posterior predictive testing, to test the absolute fit a model (Höhna et al. 2018, MBE). Moreover, this work is fundamental for any phylogenetic analysis because robust and accurate estimates of gene trees are crucial for all research areas using phylogenetics.
References:
- S Höhna, LM Coghill, GG Mount, RC Thomson and JM Brown: P3: Phylogenetic Posterior Prediction in RevBayes. Molecular Biology and Evolution, 2018, 35(4), 1028-1034
- S Höhna, MJ Landis and JP Huelsenbeck: Parallel power posterior analyses for fast computation of marginal likelihoods in phylogenetics. Bioinformatics